Types of hemophilia - Hemophilia News Today
There are several types of hemophilia, a rare bleeding disorder wherein the blood fails to clot properly. Hemophilia A and B are the most common types, while hemophilia C is comparatively rare.
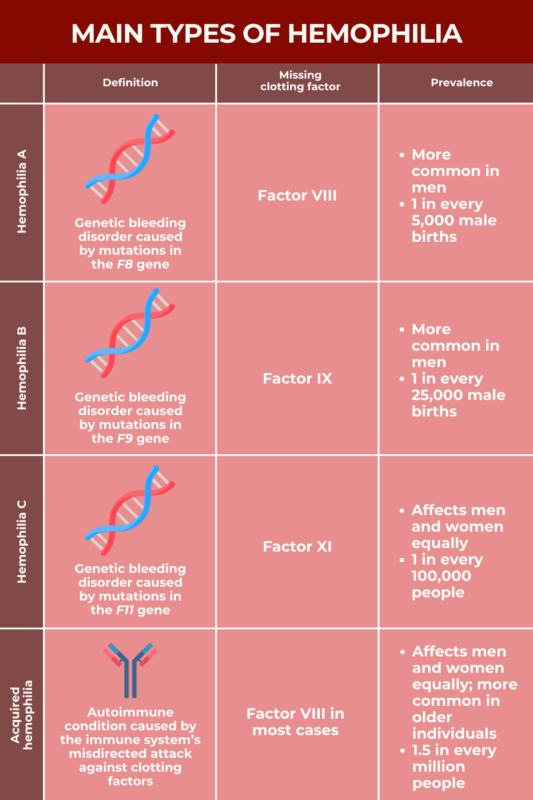
All types of hemophilia are characterized by the lack or dysfunction of blood clotting proteins called clotting factors. The type of hemophilia a patient has depends on the specific clotting factor that is missing.
Most cases of hemophilia are caused by mutations in the genes that provide instructions for making specific clotting factors, impairing the body's ability to produce a sufficient amount of functional clotting proteins. An acquired form of hemophilia occurs when the immune system accidentally attacks and destroys certain clotting proteins.
Hemophilia A
Hemophilia A, known as classical hemophilia, is caused by mutations in the F8 gene, which provides instruction for making clotting factor VIII (FVIII).
In people who don't have hemophilia, FVIII activity typically ranges between 50-150% compared with normative values. In people with mild hemophilia A, FVIII activity levels usually range between 5-40%. In moderate hemophilia A, FVIII activity levels range between 1-5%, and fall to less than 1% with severe hemophilia A.
About 60% of people with hemophilia A have the severe form of the disease. People with severe hemophilia A commonly have excessive or prolonged bleeds without any apparent cause. Those with moderate or mild hemophilia A don't typically have spontaneous bleeds, but they can experience excessive bleeding following an injury or after surgery.
Since people with more severe hemophilia tend to have more frequent bleeding problems, they are generally diagnosed earlier in life.
Hemophilia A is typically treated with replacement therapies that provide a working version of the FVIII protein that is missing, with the goal of lowering the risk of bleeds and controlling active bleeds. In recent years, gene therapies that deliver a working version of the F8 gene to cells have become available for some people.
Who develops hemophilia A?
The F8 gene is found on the X chromosome, one of the two sex-determining chromosomes. People who are biologically female have two X chromosomes. Biological males have one X and one Y chromosome.
Hemophilia A only develops if a person doesn't have a working copy of the F8 gene. Consequently, it's much more common in men, who only have one X chromosome, than in women, who have two and for whom one working copy can compensate for a mutated copy. Hemophilia A is estimated to occur in about one out of every 5,000 male births.
In about two-thirds of the cases, the mutation that causes hemophilia A is inherited from a person's parents — usually the mother, since males inherit their single copy of the X chromosome from their mother. In the remaining cases, mutations develop de novo, meaning a child has a mutation that arises spontaneously, rather than being inherited from parents.
Hemophilia A affects people of all races and ethnicities at roughly equal rates.
Hemophilia B
Hemophilia B is caused by mutations in the F9 gene, which provides instructions for making clotting factor IX (FIX). This type of hemophilia is also known as Christmas disease, after a five-year-old boy named Stephen Christmas who was the first person formally diagnosed with it in the early 1950s.
Like hemophilia A, hemophilia B can be divided into the same three categories of severity based on FIX activity levels. People with FIX activity levels ranging between 5-40% are said to have mild disease, while those with levels ranging between 1-5% have moderate disease. Those with FIX levels below 1% have severe disease. As with hemophilia A, spontaneous bleeds and joint bleeds are more common with severe hemophilia B.
Both hemophilia A and hemophilia B are marked by abnormal bleeding. While there can be a lot of variability from person to person, studies generally suggest that people with hemophilia B tend to have less severe disease than those with hemophilia A, even among patients with similar disease severity based on clotting factor levels.
Replacement therapies that deliver a working version of the FIX protein are typically used to prevent and control bleeding episodes in people with hemophilia B. Recently, gene therapies that deliver a healthy copy of the F9 gene have become available for some people.
Who develops hemophilia B?
The F9 gene is located on the sex-determining X chromosome, so, like hemophilia A, hemophilia B is much more common in men than in women. Hemophilia B is about four times less common than hemophilia A, however, with estimates indicating it occurs in approximately one in 25,000 male births.
A mutation inherited from the parents is the cause of about two-thirds of cases of hemophilia B, while the remaining one third of cases arise due to spontaneous mutations. The disease occurs at similar rates across racial and ethnic groups.
Hemophilia C
Hemophilia C, also known as Rosenthal disease, is caused by mutations in the F11 gene, which carries instructions for making clotting factor XI (FXI).
People with hemophilia C generally have symptoms that are not as severe as in hemophilia A or B. Joint and muscle bleeding also is much less common in hemophilia C than in other forms of hemophilia.
Hemophilia C often doesn't require treatment unless a patient is undergoing a surgical procedure where major bleeding is considered a risk. When treatment is needed, hemophilia C is usually managed with infusions of fresh frozen plasma (the noncellular component of blood) or with medications that slow the breakdown of blood clots, thereby helping to prevent blood loss.
Who develops hemophilia C?
Unlike the F8 and F9 genes, the F11 gene isn't located on a sex-determining chromosome. Therefore, every person inherits two copies of the F11 gene, one from each parent. Severe hemophilia C will usually only develop if a person has mutations in both F11 copies. A person who carries only one mutated copy generally won't have severe symptoms, but may still pass on the mutated gene to their children.
If two people who are both carriers of a faulty F11 gene copy have a child, there is a:
- 25% chance the child will inherit both mutated copies of the F11 gene and develop hemophilia C
- 50% chance the child will inherit one mutated gene copy and be a carrier
- 25% chance the child will not inherit any mutated copies of the gene and be healthy.
In some instances, just one mutated gene copy is sufficient to cause hemophilia C, though this is rare and usually doesn't lead to severe disease. If a person with hemophilia C caused by one mutated copy of the F11 gene has children, there's a 50% chance of passing the disease-causing mutation to their biological children.
Hemophilia C is notably more rare than hemophilia A or B, affecting about one out of every 100,000 people. Unlike other forms of hemophilia more commonly seen in men, hemophilia C affects men and women equally. Hemophilia C is also more common among Ashkenazi Jews, affecting about 8% of people in this ethnic group.
Acquired hemophilia
While most types of hemophilia are genetic disorders caused by mutations that a person is born with, acquired hemophilia is an autoimmune disease that develops over a person's lifetime.
In acquired hemophilia, the immune system system fails to recognize a clotting factor as a healthy part of the body. As a consequence, immune cells produce antibodies that attack the clotting protein as if it were an infectious agent, preventing it from functioning normally.
In most cases of acquired hemophilia, the immune system specifically attacks FVIII, the clotting protein that's missing or is defective in hemophilia A. More rarely, acquired hemophilia also can develop due to an autoimmune attack targeting other clotting proteins.
The severity of acquired hemophilia can vary widely. Around one in three people with acquired hemophilia have relatively mild symptoms and don't require specific treatment, but some patients experience frequent and serious bleeds, which can be life-threatening, especially if the condition isn't diagnosed correctly and treated promptly.
Because it's so rare, there aren't well-established guidelines for treating acquired hemophilia, so treatments tend to be highly tailored to the individual patient. Broadly, treating acquired hemophilia has two goals — control bleeding and reduce the production of antibodies that target clotting factors.
Who develops acquired hemophilia?
Acquired hemophilia is very rare, affecting an estimated 1.5 out of every 1 million people. The disorder occurs at similar rates in men and women, and, although it can develop at any age, it most commonly occurs in the latter decades of life. It's estimated that about 80% of people with acquired hemophilia are older than 65.
In about half of the cases, acquired hemophilia develops in people who already have other underlying health problems, such as:
- autoimmune diseases like lupus, rheumatoid arthritis, myasthenia gravis, or multiple sclerosis
- cancer, including lung, prostate, pancreas, breast, and several types of blood cancers
- skin conditions, such as psoriasis, pemphigus, and epidermolysis bullosa
- infections diseases like hepatitis B and C
- inflammatory bowel disease
- transplant-related disorders like chronic graft versus host disease
- asthma or chronic obstructive pulmonary disease (COPD).
Acquired hemophilia also can develop as a complication following childbirth, which accounts for nearly 10% of cases. It also may develop as a reaction to blood transfusions or to certain medications, including some antibiotics and non-steroidal anti-inflammatory drugs (NSAIDs).
No specific cause or underlying health condition can be identified in about half the patients, however.
Hemophilia News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Comments
Post a Comment